(EU)2017/745和(EU)2017/746这两项关于医疗器械和体外诊断试剂的法规分别于2021年5月26日和2022年5月26日起全面实施。它们的目标是改进医疗器械质量、提高安全性和可靠性,增强为患者提供透明性和信息,提高警戒和市场监督。这两项法规虽然都规定了过渡期,但在此期间,符合先前指令的器械仍然可以投放在欧盟市场上。为了确保平稳过渡,欧盟必须定期了解实地情况,并收集有关利益相关方(包括公告机构、经济运营商、提供保健服务者以及患者代表等)目前开展的活动的具体数据。
于是欧盟委员会健康与食品安全总局(DG SANTE)通过欧洲健康与数字执行机构(HaDEA)委托开展一项名为’支持监测欧盟市场上医疗器械供应情况的研究“,该研究将从2022年12月开始,持续36个月,研究检查医疗器械法规实施的情况,并有助于确定需要应对的潜在挑战以及可能的解决方案。
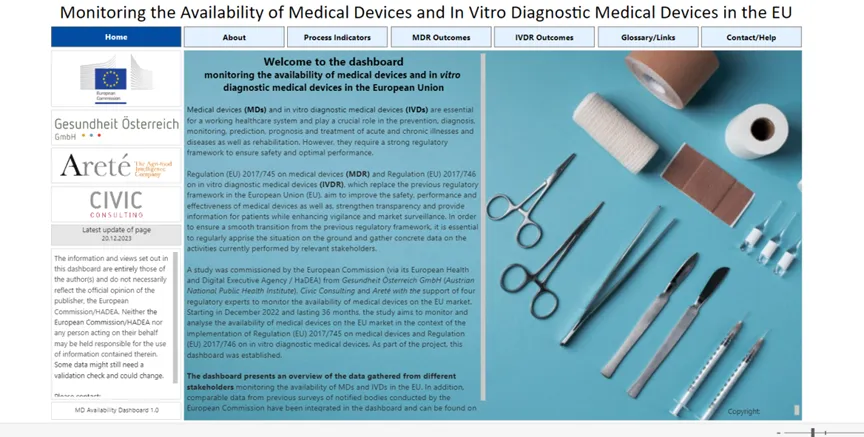
研究小组还设计了一个“Dashboard”,其中包含来自利益相关者调查的汇总数据,并定期更新,目前有39家MDR公告机构和10家IVDR公告机构参与了本研究,从“Dashboard”中可以得到不同周期MDR/IVDR证书的申请情况,质量体系证书的颁发情况,制造商与公告机构之间的签约情况,拒接理由,附录XVI非医疗用途的器械申请和认证情况等诸多信息。
值得注意的是,申请和签署的公告机构协议是有资格获得CE证书延期的必要条件。指南MDCG 2022-11 Rev.1中也有提及由于MDR/IVDR下的文件审核较为严格,大多数制造商的申请不完整,这也导致了认证过程的延误。因此制造商应充分利用好法规的过渡时间并及时提交符合性评估申请。不要等到申请的最后一刻,冒着没有及时转移到MDR/IVDR的风险,失去欧盟市场的准入机会。
关于医疗器械和体外诊断试剂的过渡期的更多信息可以查看MDCG 2022-18,以及法案(EU)2023/607,根据MDR第120(3c)条(e)点,制造商或授权代表必须在2024年5月26日之前根据MDR附录VII第4.3节第一小节的规定提交正式的合格评定申请。而公告机构必须在2024年9月26日之前根据MDR附录VII第4.3节第二小段签署书面协议,才能从延长的过渡期中受益。
意义:制造商可以根据自身产品的申报情况查看不同界面模块的显示结果,了解自身产品的注册流程和周期,并依照法规要求,理解不同审核下的欧盟文化和差异,澄清问题,及时沟通,审评文件细致,一次就做好应对。
更多的问题欢迎咨询丽和康,祝您在法规上顺利前行!
原文链接:
https://health.ec.europa.eu/study-supporting-monitoring-availability-medical-devices-eu-market_en


