“过渡期”一直以来都是CE注册所关注的重点话题。随着(EU)2023/607的正式发布,关于MDR过渡期的时间,欧盟也给出的明确的规定,如下图所示:
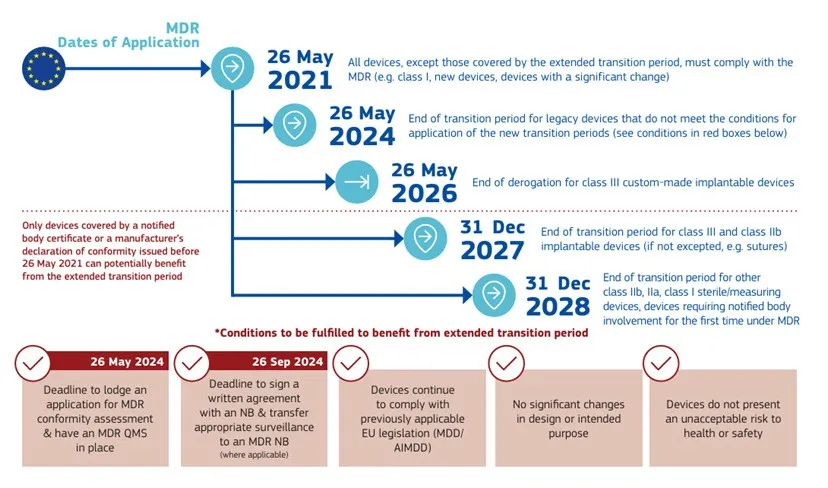
2024年的5月26日,是MDR过渡期延期的一个关键节点,与此同时当地时间5月27日,欧盟发布了“ MDCG 2022-4 Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR with regard to devices covered by certificates according to the MDD or the AIMDD ” 指南的第2次修订。本次修订对指南的部分内容进行了调整,使其更符合(EU) 2023/607以及(EU) 2017/745以及(EU) 2017/746过渡条款的要求。
本指南适用于公告机构对MDD或AIMDD遗留器械过渡期的监督审核,文件还涵盖了有关某些制造商义务的要求,尤其是其质量管理体系的要求。
• 对制造商质量管理体系的要求及相关义务
1. 不迟于2024年5月26日应根据MDR建立了质量管理体系。此外,自MDR申请之日起,关于经济运营商的注册登记和设备的上市后监督、市场监管、警戒等所有相关要求适用于“遗留设备”;
2. 在强制使用Eudamed数据库或其相关模块之前,制造商或其授权代表应适用各自的国家规定,并考虑指南MDCG 2021-1 Rev.1;
3. 基于MDD下的分类,“遗留设备”也应符合MDR中关于PMS以及PSUR规定的要求。在过渡期内,不应考虑MDR下其风险等级的可能变化。为了符合MDR的要求,AIMDD下的有源植入式器械应被视为III类器械;
4. 根据MDR起草PSUR,且PSUR必须向主管当局提供;
5. 根据MDR第120(3e)条,与上市后监督、市场监管、警戒、经济运营商和设备注册无关的MDR要求不受监督。
• 对质量管理体系文件的审查重点
1. MDD或AIMDD证书所覆盖的设备范围保留或发生更改;
2. 哪些设备已停止使用或不包括在应用程序中。这还包括需要考虑那些打算替代遗留设备的设备;
3. 制造商的MDR的过渡计划;
4. 对于调整了的质量管理体系,需要考虑到指南MDCG 2020-3 Rev.1;
5. 如果制造商对上市后监督、市场监管、警戒、经济运营商和设备注册的质量管理体系进行了必要的调整。这可以通过验证制造商已经改变了上市后监督等程序来实现;
6. 关于新的上市后监督(PMS)要求:
• 与上市后监督相关的所有适当过程,包括风险管理和临床数据,都应纳入PMS plan;
• 如果所有上市后监督活动的输出都包含并反映在PSUR中,并且在适用时,PSUR更新周期是适当的,并且符合MDR第86条中规定的风险等级。
公告机构在过渡期内会侧重以上条款对制造商的质量体系进行审核,如果过渡期不再适用,公告机构应考虑对MDD或AIMDD证书的任何影响。如果审核发现重大不符合项,可能对患者、使用者或其他人的健康或安全构成不可接受的风险,则公告机构需要采取行动,即暂停、限制或撤销证书,并通知相关主管部门。
根据MDR的规定,颁发MDD或AIMDD证书的公告机构应继续负责有关其已认证的设备的适当监管要求。其对适当监管的责任最迟将于2024年9月25日结束。不迟于2024年9月26日,制造商应于公告机构签署MDR认证的书面协议,届时公告机构对申请所涵盖的遗留设备进行适当的监督。
值得注意的是,制造商只有在规定时间内建立MDR下的质量体系以及与公告机构签署MDR认证的书面协议才能享受到过渡期延长的红利。直到今日,关于欧盟要求建立MDR下质量体系的时间(2024年5月26日)已经过去了,还没有按要求进行的企业应抓紧时间。丽和康拥有法规认证的顶尖团队,致力于提供多国医疗器械注册全流程服务、还可提供质量体系建立,运行辅导以及模拟审核等多项服务,有需要的朋友,欢迎与我们联系。
原文链接:
https://health.ec.europa.eu/latest-updates/revision-2-mdcg-2022-4-mdr-appropriate-surveillance-2024-05-27_en


